Chronic Myeloid Leukemia, BCR-ABL1 positive
Clinical features
- Incidence of 1-2 cases/100k population.
- Acute radiation may be a risk factor, but unlike other MPNs, there is little inherited 3predisposition.
- Insidious onset. Most are either asymptomatic (~50%), or present with some combination of B symptoms and palpable splenomegaly (~50%).
- ~5% are diagnosed in blast phase.
- Natural history without treatment is progression to accelerated/blast phase in 3-5 yrs.
Morphologic features
- Peripheral blood:
- Leukocytosis with myelocyte bulge, basophilia, and eosinophilia. Children often have higher WBC counts than adults.
- Dysplasia is absent.
- Bone marrow:
- Although diagnosis can be made on peripheral blood, bone marrow aspiration is essential for confirming the phase of disease. Biopsy is not necessary but is recommended.
- Hypercellular marrow with granulocytic hyperplasia and left shift.
- Small “Dwarf” megakaryocytes with hyposegmented nuclei; many cases also show megakaryocytic proliferation.
- Increased eosinophils and basophils.
- Pseudo-Gaucher cells (non-specific).
- Moderate to marked reticulin fibrosis may be present and is often associated with splenomegaly and clustered small megakaryocytes.
Disease phases and progression
| Criteria | Additional features | |
| Chronic phase (CP) | Does not meet criteria for accelerated or blast phase. | Blasts are usually <5%. |
| Accelerated phase (AP) | Any of the following, despite therapy:
– Persistent/increasing WBC count (>10x10E9/L). – Persistent/increasing splenomegaly. – Persistent thrombocytosis (>1000x10E9/L), or thrombocytopenia (<100x10E9/L) not due to therapy. – ≥20% basophils in peripheral blood. – 10-19% blasts in blood and/or bone marrow. – Evidence of an additional clonal cytogenetic abnormality in Ph+ cells (either present at diagnosis, or which arises during therapy). |
Provisional response-to-therapy criteria for AP include resistance to 1st TKI, any resistance to 2 sequential TKIs, and ≥2 BCR-ABL mutations. |
| Bast phase (BP) | – ≥20% blasts per WHO (some guidelines use ≥ 30%) in blood or bone marrow.
– Extramedullary blast proliferation. |
Most cases have myeloid lineage; 20-30% of cases show lymphoid lineage (usually B-cell).
The finding of any number of lymphoid blasts should raise concern, and focal larger sheets of blasts in bone marrow can be considered equivalent to BP. |
Immunophenotype
- Main role of immunophenotyping is determination of blast lineage.
- Expression of CD7 by myeloid blasts may have poor prognosis.
- Most lymphoid BP cases are pre-B, with expression of TdT.
- Expression of lymphoid antigens in myeloid BP, and vice versa, is common.
Genetic Features
- 90-95% have t(9;22)(q34.1;q11.2) resulting in the Philadelphia chromosome (der(22)) and the formation of BCR-ABL1 fusion gene and fusion protein with constitutive tyrosine kinase activity.
- Remaining cases have variant or cryptic translocation.
- BCR-ABL1 breakpoints/isoforms:
- The major breakpoint (p210 isoform) is seen in most cases
- The minor breakpoint (p190 isoform) is seen in small amounts in most cases of CML, and is sometimes associated with monocytosis. This is the isoform most often present in Ph+ B-ALL.
- The p230 isoform is associated with neutrophilia and thrombocytosis.
- Mutations in the BCR-ABL1 kinase domain can lead to resistance to 1st generation TKIs, less so to 2nd generations.
Therapy, prognosis, and outcome
- Treated with 1st generation TKIs (imatinib) and 2nd generation TKIs (nilotinib, dasatinib, bosutinib, ponatinib).
- The most important prognostic factor is response to therapy.
- 10-year overall survival has improved to 80-90% with the advent of TKIs; approaching that of the normal population.
References (APA style)
Video
Images
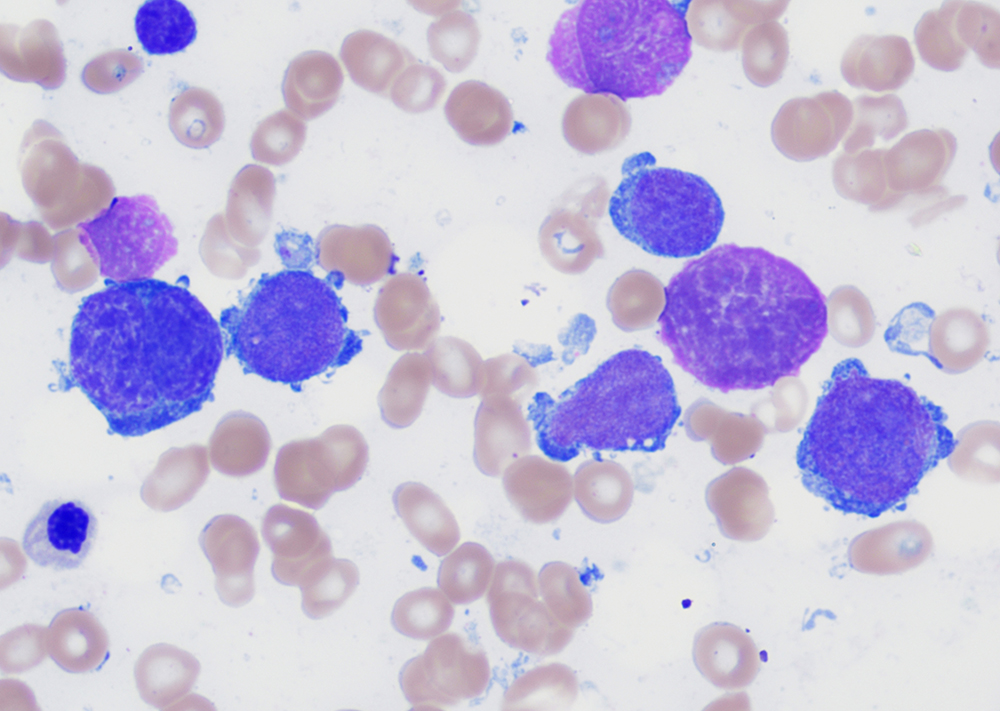
Image Title
Sed ut perspiciatis unde omnis iste natus error sit voluptatem accusantium doloremque laudantium, totam rem aperiam, eaque ipsa quae ab illo inventore
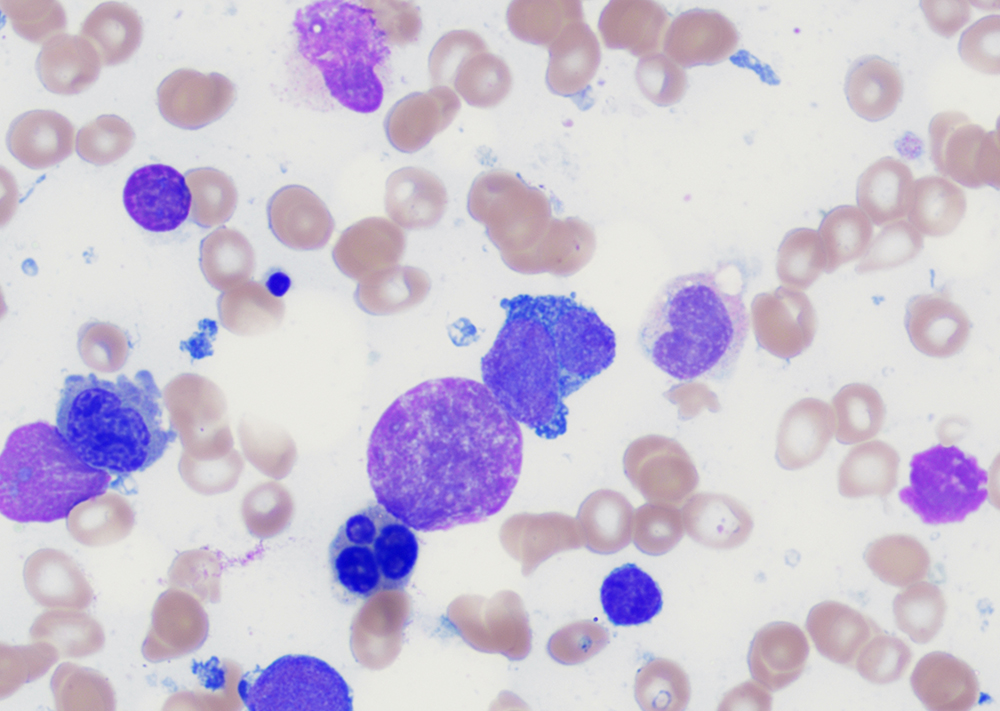
Image Title 2
Sed ut perspiciatis unde omnis iste natus error sit voluptatem accusantium doloremque laudantium, totam rem aperiam, eaque ipsa quae ab illo inventore
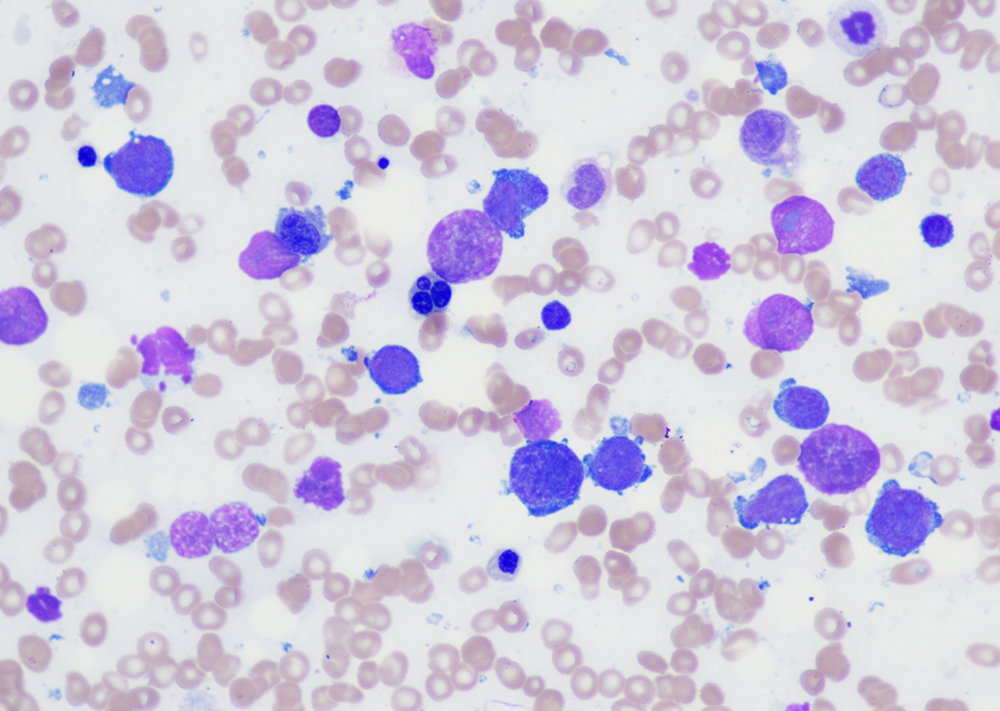
Image Title 3
Sed ut perspiciatis unde omnis iste natus error sit voluptatem accusantium doloremque laudantium, totam rem aperiam, eaque ipsa quae ab illo inventore
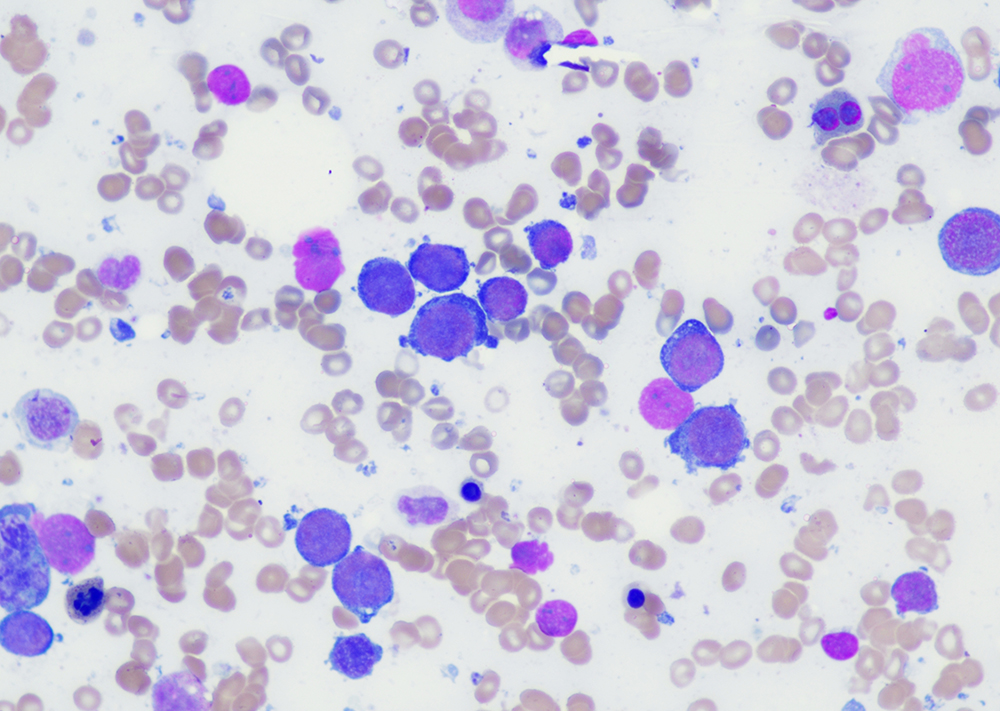
Image Title 4
Sed ut perspiciatis unde omnis iste natus error sit voluptatem accusantium doloremque laudantium, totam rem aperiam, eaque ipsa quae ab illo inventore
Image Title 5
Sed ut perspiciatis unde omnis iste natus error sit voluptatem accusantium doloremque laudantium, totam rem aperiam, eaque ipsa quae ab illo inventore